This page provides some basic information on manipulating the fission yeast genome by constructing gene disruptions or targeted integrations, using one-step or two-step integration protocols. Check our methods page for more protocols. If you need more basic information about yeast genetics, try this yeast genetics course site. You might also check out the Nasmyth lab’s page, How to delete a pombe gene in 8 days.
Disruptions
Gene disruptions allow the investigator to create a null mutation in the gene of interest, which is usually replaced with a marker (mkr+) to be followed genetically. the basic strategy is to isolate a fragment of DNA containing the marker and flanking sequence homology to the gene of interest (your favorite gene, or yfg1+). The ends of the fragment act as double strand breaks to invade the homology region, resulting in a double crossover that replaces the target gene in the chromosome with the fragment. Importantly, the only sequence on the fragment is the marker and the homology domain; there is no plasmid DNA and no ars element.
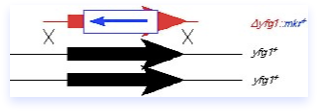
Gene disruptions usually require the cloned gene of interest to be manipulated in vitro. Insertion-disruptions are more common in the older literature. In these constructs, a marker is inserted into a restriction site within the cloned sequence. The resulting construct contains an interrupted gene. However, gene fragments could still be synthesized from the intact portion of the gene, potentially clouding the analysis. More commonly, deletion-disruptions are used that delete a central portion of the open reading frame, or the entire ORF.

Recently, PCR-mediated disruption methods have become common, in which long primers providing the homology are used to amplify a marker from a plasmid, as diagrammed below. However, fission yeast has a high rate of non-homologous recombination, and homologies of less than 200 bp can target inefficiently, leading to a high background of random insertions. This has led to some refinement of the original method, designed to increase the size of the homologous region.
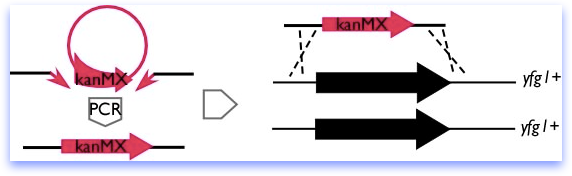
In the Forsburg lab, we generally use a combination of methods and try for at least 200bp homology on each side of the marker. Our constructs are specific to the ORF; we have observed cases where adjacent genes are affected even when only a few bases outside the ORF are deleted. While rare, this is extremely annoying, and we prefer to be safe-rather-than-sorry.
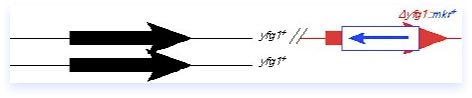
The key in gene disruption is in analysis of the product. As shown in the diagrams, generally we disrupt genes in diploids. If the gene is essential for viability, and is disrupted correctly, then upon random spore analysis, no viable haploids containing the disruption marker will be isolated. Alternatively, you can do tetrad analaysis. Analysis of the disruptant’s phenotype can be carried out by bulk spore germination in liquid media. If the gene is not essential, the mutation may still confer a phenotype, which will be tightly linked to the disruption marker.
Of course, the construct may integrate into the wrong locus (as shown in the diagram at right), and even generate a phenotype. This must be distinguished from the correct construct. Many people use colony PCR, but requires careful primer design. Southern blotting can be very convincing, but you must be careful to use a probe that hybridizes to sequences OUTSIDE the disruption construct. A low-tech, but convincing way to prove you have disrupted the correct gene is plasmid complementation of the disruption phenotype, assuming it has one. You transform with disrupted diploid with a plasmid containing the intact gene and a different marker; upon sporulation and random spore analysis, you determine whether or not you can recover both the disruption marker AND the plasmid marker in wild type haploid offspring. We know of several investigators who were persuaded by their Southern or PCR but proven wrong by the biology, so this control is absolutely required in the Forsburg laboratory in addition to molecular characterization.
Once-step transplacement can also be used to insert a marker downstream of a gene, to replace one allele with another, or to carry out other manipulations. The methods used depend upon the nature of the markers and whether they are counterselectable.
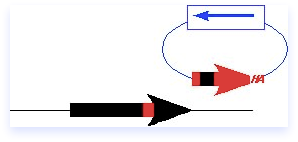
Integrations
Not all integrations into the genome are disruptions. Insertions or epitope tags may also be constructed. Such tags can also be inserted into yfg1+ using a very short transplacement of the C-terminus of the gene with a tagging construct, usually generated by PCR with long oligos. As described above, efficiency of this method is variable locus to locus and improved by increasing the amount of homology (i.e, using longer oligos to PCR the tag). The methodology and caveats are similar to those described above and there are specially built vectors for this purpose. The advantages to this approach is that it’s fast, and there is no excess DNA integrated. The disadvantages is that some regions of the genome are reluctant to be modified in this way.
Although more classic methods of manipulation of the genome are less common now, the principles are important because they can still be useful for gene replacements and other modifications. The most classic verions Campbell-model integration to make an insertion; it is a rapid method to epitope tag yfg1+ but can also be used to create two-step gene disruptions or transplacements via a loop-in loop-out method.
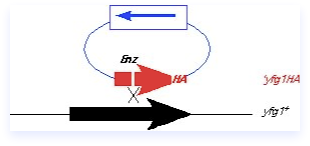
The integrating fragment in this case is actually a plasmid containing a yeast marker, but no yeast origin. It contains a C-terminal fragment of yfg1+, including an epitope tag, but no promoter or ATG. Thus, this fragment cannot be expressed, and should not complement a mutant. There is also a unique restriction site (Enz) within the fragment that is cleaved prior to transformation, so that the plasmid is linearized. The ends of the fragment are invasive and help target to the homologous region on the chromosome. A single crossover occurs.
The integrated construct contains a partial tandem duplication in which the manipulated C-terminus of the gene of interest is fused to the endogenous locus, and the non-expressed C terminal fragment is integrated downstream. The construct can be screened by looking for expression of the tagged protein (only the correct construct should give a protein of the correct size), and the tight linkage of the marker to the locus under study. Although many genes are amenable to being tagged in this way, epitope tagging can also induce phenotypes including cold- or heat-sensitivity or synthetic genetic interactions, so some caution is required. It’s a good idea to make the construction in a diploid first (as diagrammed in this panel), to avoid any selective pressures, and then isolate the haploid. If the tag doesn’t cause any deleterious phenotypes, the integration will be quite stable even in the absence of selection. However, if the tagged protein is not fully functional, there may be selective pressure leading to marker loss if selection is not maintained.
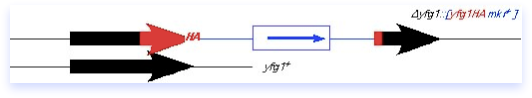
What goes in can come out, and may result in a replacement of plasmid-for-chromosomal DNA, as diagrammed here. The plasmid can loop out by the same sort of mechanism it used to integrate, although importantly the crossover may not occur at the same place. If there were a mutation elsewhere in the red yfg1 fragment, you can see that it might be possible to leave that behind in the genome. The loop-out method can therefore be adapted to integrate disruptions or new alleles into the genome. It is also possible to use this method in conjunction with a typical disruption construct (such as diagrammed in the preceding section) to carry out a replacement of the null allele with a different allele, for example a temperature sensitive mutation. One must remain aware however that not all crossovers are equal: the power of selective pressure can strongly influence the direction of the integration, and therefore one must rigorously verify the correct structure of the resulting chromosome as described in the preceding section.