Solar Energy Conversion: From Solar Fuels to Organic Photovoltaics and Nanomaterials
Solar fuels are attractive due to their renewable nature and environmental benefits and
complement stored electricity solutions. Understanding the mechanism involved in the initial steps involved in the photocatalytic system is vital to designing new sustainable photocatalytic systems. Photosensitizer is the part of the photocatalytic system that absorbs light and drives the charge generation process. Current state-of-the-art photosensitizers are not efficient enough and are not scalable due to the involvement of precious elements like Ru and Ir. In this project, we investigate the excited state dynamics of photosensitizers that are designed based on earth- abundant elements like Cu, Zn, and Zr. A wide range of spectroscopic tools are employed to obtain a complete picture of the initial steps involved in these molecules once they absorb photons. This includes transient absorption spectroscopy in the picosecond and nanosecond time regimes, time-correlated single photon counting, and other complementary steady-state
spectroscopic and electrochemical techniques.
Organic photovoltaics stand apart from their inorganic counterparts due to their flexibility, lightweight character, low cost, and transparency. While organic photovoltaics based on small molecules have manifested a competing power conversion efficiency of 19%, the practical application of these systems is limited because of their synthetic complexity and expensive large- scale production. A promising alternative is small molecules with a BODIPY core. However, the highest efficiency of these molecules lags by 10 % of the state-of-the-art molecules used in organic photovoltaics. One approach to dramatically increase the efficiency of these molecules is to find exciton recombination pathways and suppress them, wherever possible. This project aims to investigate the mechanism of exciton formation and its dynamics in organic molecules employing time-resolved spectroscopic techniques. In addition, novel nanomaterials are also investigated for applications in solar energy conversion.
Student-in-chief: Thabassum Nattikalungal – tnk (at) usc (dot) edu

Excited State Photodynamics of Heteroaromatics in Bulk and Surface Aqueous Phase
Water is crucial for every aspect of human life, from biology to the environment. It is the predominant medium in which the chemistry of life takes place. Yet even today in this world of artificial intelligence, scientists are still struggling to understand the intricate details of how water molecules interact with each other or with dissolved solutes in aqueous solutions. Specifically, complex interactions of water with small aromatic bio-chromophores that govern almost all aspects of the natural functioning of the human body, plants and the global climate are still a topic of intriguing debate. The bio-chromophores in presence of water, ubiquitous in nature, are naturally exposed to the sun’s UV rays, every day, and undergo complex photochemical reactions. They also end up in atmospheric organic aerosols that potentially possess risks involving human health and environmental pollutants. Their photochemical dynamics and fate in bulk aqueous phase as well as air-water interface become environmentally and biologically relevant.
Naturally occurring phenols, substituted phenols and indoles are crucial molecular motifs in bio- chromophores like the non-essential amino acid tyrosine or the essential amino acid tryptophan. Elucidating key differences in their photodynamics in the bulk versus interface drives our experimental interest. Lack of complete photochemical information of a species in the bulk aqueous phase hinders full characterization of surface specific properties. In Bradforth lab, we exploit home-built state-of-the-art spectroscopic techniques like femtosecond to microsecond broadband polarization-dependent transient absorption (one-photon/1PA) spectroscopy and broadband two-photon absorption (2PA) spectroscopy to unravel key features about the photoexcited molecular electronic structure and dynamics in bulk. A comparative study of these transient absorption spectroscopies with our time-resolved liquid-jet photoelectron spectroscopic (LJPES) measurements could finally reveal the similarities and differences between bulk and surface photochemistry in these molecules.
Our investigation of the photoionization dynamics of indole in aqueous solution show remarkable similarities to a solvent-exposed tryptophan residue in a protein (NCp7), indicating that our detailed mechanism is likely the most appropriate for describing photodynamics of tryptophan in its native environment (1). Our investigations of bulk-sensitive 2PA spectra of water (2), methanol (3), ethanol (3), iodide (4), liquid alkanes (5) and phenol (6) have provided crucial information about excited state symmetry that aids in the interpretation of surface-sensitive E-SFG (electronic-sum frequency generation) spectra. Bradforth group and collaborators (8) have also published articles that provide insights into the significance of UV- LJPES, specifically in aqueous phase, as both a surface-sensitive and a bulk-sensitive tool. Experimental observations from 2PA, combined with 1PA, surface specific E-SFG and UV-LJPES can help decipher unresolved key features and reveal new details about the excited molecular electronic structure at bulk and aqueous interface.
(References:
1. Kumar et al, J. Phys. Chem. B, (2024); PMID: 38655896.
2. Elles et al, J. Chem. Phys. (2009) 130, 084501.
3. Bhattacharyya et al, J Phys Chem A. (2019) 11;123(27):5789-5804.
4. Bhattacharyya et al, J Phys Chem Lett. (2020);11(5):1656-1661,2020 Feb 14. PMID: 32040333.
5 Bhattacharyya et al, J. Phys. Chem. A 2021, 125, 36, 7988–7999.
6. ACS poster.
7. Seidel R, Winter B, Bradforth SE. Annu Rev Phys Chem. 2016 May 27;67:283-305)
Student-In-Chief: Shivalee Dey Email: shivaled@usc.edu

Electronic Structure of Aromatic Anions in Ethereal Solvents
Liquid ammonia is a useful solvent in organic chemistry for its ability to stabilize carbanions. These carbanions are typically generated by adding sodium metal to liquid ammonia, which produces solvated electrons which then add to pi-conjugated species. Despite this often-used reagent, the mechanism of action—the principle behind how this reaction works—isn’t very well known.
Structurally, there are different motifs of the solvated electrons which depend on the concentration of alkali metal added. At dilute concentrations of 1 micromolar, the solvated electrons exist as discrete doublet species. At higher concentrations ~0.1 M, the electrons spin-pair to form the mysterious solvated dielectron. Higher concentrations afford a visually striking transition to the metallic state (Mott transition).
Can we measure the binding energies of these different species to understand how they form? Our preliminary work suggest yes, having created the first liquid microjet of neat cryogenic ammonia in vacuum to measure the X-ray photoelectron spectrum of liquid ammonia with our collaborators at BESSY II in Berlin. We have replicated this success with a liquid ammonia sampling system of our very own at USC for time-resolved studies. Work on the alkali metal solutions is continuing both at home and abroad, with the later aim of studying the electronic structure of carbanions otherwise inaccessible in the gas-phase.
Student-in-chief: Brandon Young – bmyoung (at) usc (dot) edu

The Role of Flanking Bases in DNA Photodamage
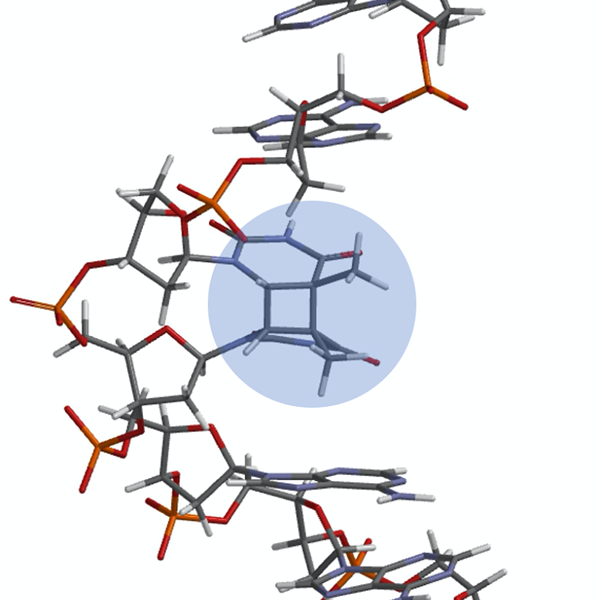
Light from the sun provides energy to sustain life on Earth. Unfortunately, it can also harm those same living organisms that seek the warmth of the day. A prevalent source of damage to DNA strands is from UV irradiation. Absorption of UV light can lead to the [2+2] cycloaddition of two neighboring thymines or cytosines, thus resulting in the formation of the cyclobutane pyrimidine dimers (CPDs). This is a common lesion in DNA which, upon replication, can lead to mutagenic and carcinogenic effects. Biophysical research on DNA damage by UV radiation often focuses on the role that primary structure plays in the formation of photoproducts. Although it is well known that the electronic excited states of nucleic acids are highly stable, less is known about the excited states dynamics of the oligomer or the role of secondary structure in the formation of CPDs. Previous investigations have demonstrated that bases adjacent to thymine affect the photochemical yield for the CPD formation. However, the role flanking bases have on the rate of CPD formation has yet to be determined.
The formation of CPDs occurs through two different pathways, depending on the stacking of the bases in the strand. When the bases are stacked and in a reactive configuration, the dimerization occurs through an ultrafast, singlet pathway. When the bases are not stacked and are free to move around, this allows the system to transition to a triplet state from which the CPD is formed. It has also been proposed that the presence of charge transfer (CT) states have an important role in regulating this latter dimerization process by either enhancing or impeding it.
The goal of this project is to initially characterize spectroscopic signatures of the CT state, the approach to the conical intersection that leads to dimerization, and the final CPD photoproduct. Transient absorption (TA) will be used to perform characterization of some of the spectroscopic signatures. Meanwhile, those signals not present in TA will be identified using femtosecond stimulated Raman spectroscopy. These experiments will be performed in different sequences to assess the role flanking bases and, in general, secondary structure have on the rates of CPD formation
As an NIH fellow, José seeks to add a new biological perspective on how a simple photochemical transformation in DNA can have a deeper impact on genome stability. When isolated, the CPD photoproduct has limited power to harm the genome. However, in regions of DNA called ‘satellites’, which are known for many instances of AA repetitive sequences, the presence of TT dimers is more profound. As part of a chemistry-biology interface in Dr. Irene Chiolo’s lab, he will study the accumulation of UV photodamage in satellite DNA.
Student-in-chief: José L. Godínez Castellanos
Photo: X-ray crystallographic depiction of the thymine CPD (highlighted in blue) in the complimentary strand of the DNA dodecamer duplex. McAteer, K., Jing, Y., Kao, J., Taylor, J.S., Kennedy, M.A. (1998) J Mol Biol 282: 1013-1032