1. The name is Bond: Hydrogen Bond
Starting in the early 1920s there has been continued interest in understanding the nature and dynamics of hydrogen-bonded networks in diverse environments. In the gas phase, emphasis has been placed on studies of clusters in the cold environment of supersonic expansions, with most reports focused on spectroscopy. The state-specific energy flow in hydrogen bonded networks and the vibrational predissociation (VP) dynamics, which are the focus of our work, are much less explored experimentally and are still poorly understood. At the same time, because of the relevance of H-bonded networks to environments ranging from biological molecules in cells to gaseous species erupting from icy bodies in the solar system, theory groups are redoubling their efforts towards generating accurate potential energy surfaces (PES’s) for molecules such as water and ammonia, which will describe the behavior of isolated molecules, clusters, liquids and ices.
Our own interest in the predissociation of weakly-bound molecules derives from our work on the photodissociation dynamics of molecules and radicals whose excited states are weakly but covalently bound. This work has led to studies of molecules that are weakly covalently bound in both their ground and excited states, and our work on the NO dimer (more correctly cis-ONNO), which is bound in its ground state by 700 cm-1, is a good example. Such weakly bound molecules are important in the cold environment of the upper atmosphere and in interstellar space.
We realize that very little is presently known about the VP dynamics of simple H-bonded dimers and trimers of polyatomic molecules. Surprisingly, even the bond dissociation energies of dimers that have become benchmarks for theory [e.g. (H2O)2, (NH3)2, and HCl-H2O] have not been measured. So far dynamical calculations on accurate PES’s that describe VP have been carried out only for the simplest dimers with an atom or a diatom as a subunit. Our group now collaborates with the theory groups of Prof. Joel Bowman at Emory University and Prof. Tony McCaffery at Sussex University, UK.
The attractive forces that hold H-bonded clusters together give rise to distinct fingerprints in vibrational energy flow patterns and VP dynamics. Energy disposal in VP reflects the extent of vibrational couplings within each subunit, with the other subunits, and with intermolecular modes. Because of the disparity between the frequencies of inter- and intramolecular vibrational modes, IVR is often incomplete, giving rise to nonstatistical predissociation and state-specificity in fragments’ rovibrational populations.
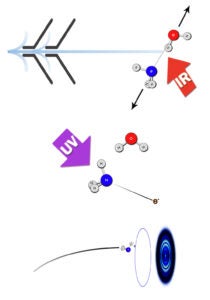
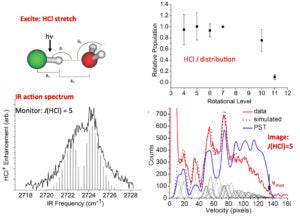
The imaging methodology we have developed for these studies is unique in its ability to provide accurate dissociation energies, D0 (~ 10 cm-1 accuracy), as well as fragment rovibrational energy distributions that reflect energy transfer across the hydrogen bond and within the dimer subunits. Moreover, it provides pair-correlated energy distributions, i.e. the rovibrational energy distribution in one fragment correlated with a selected quantum state of the other. We are not aware of any other method that gives pair-correlated distributions of molecular fragments of VP with better state selection. So far, we have studied successfully the predissociation of the following dimers: C2H2-HCl, C2H2-NH3, H2O-HCl, and H2O-NH3, as well as (H2O)2 and (D2O)2.
A very recent success is our ability to probe state-specifically for the first time water fragments from the predissociation of a cluster. Previously we were able to monitor the HCl product in the VP of H2O-HCl, and now we can determine directly the energy distribution in the water fragment as well and obtain images of selected water levels. This is a crucial step towards achieving our goal to investigate the VP of the water dimer.
We are continuing our studies of VP of polyatomic dimers and plan to extend them to the VP of cyclic trimers, which would serve as benchmark cases of energy transfer in H-bonded networks. We will determine bond dissociation energies as well as detailed energy flow patterns into fragment rotational, vibrational and translational degrees of freedom at the pair-correlated level. Because of the transformative nature of this research to several scientific communities, we believe that these challenging experiments are well worth the effort.
Specific systems include:
* H(D)Cl-water dimers and mixed cyclic trimers. The complexes of this strong acid with water would serve as examples of H-bonded interactions in which incipient proton transfer should play a role in energy transfer and VP.
* Water and ammonia dimers and trimers. These complexes serve as benchmarks for testing high-level PES’s and for dynamical calculations. We can easily monitor ammomia state-specifically and now we can also monitor water fragments.
* Mixed ammonia-water trimers. The binding energies in these mixed trimers are larger than in (NH3)3 and (H2O)3. The results will be compared with high-level dynamical calculations.
There is no need to expand on the importance of H-bonding in water in our solar system but water clusters are important not only in isolation. They also attach to positive and negative ions and interact with them. Of special interest is the use of size-selected water and ammonia clusters as “nanowires” through which H atoms are transferred to transform keto to enol tautomers. Ammonia is of fundamental interest as a Lewis base, a solvent, and a constituent in solar icy bodies. Mixtures of ammonia and water are relevant to chemical evolution, as a component of the surfaces of Neptune and Uranus, in atmospheric chemistry, and in gas eruptions detected by the Gemini telescope and the Cassini mission.
2. Photoinitiated reactions of radicals and diradicals in molecular beams
Open shell species such as radicals and diradicals are central to reactive processes in combustion and atmospheric chemistry. Our group is engaged in a long-term program to study several intriguing species, such as hydroxyalkyl radicals and methylene, the prototypical carbene. Our goal is to investigate the dissociation and ionization dynamics of radicals and diradicals for which multiple dissociation pathways, including molecular rearrangements, compete. These studies offer opportunities to address fundamental issues related to their open shell nature and the multitude of nonadiabatic interactions and product channels that characterize these systems.
The experimental approaches that we use are: (i) preparation of radicals by pyrolysis, photolysis and chemical reactions; (ii) diagnostics of reactants and products by REMPI and VUV photoionization; (iii) laser excitation schemes such as double resonance overtone pumping to reach vibrational levels above the dissociation barrier, UV and IR+UV excitation to reach dissociative states or ionization, etc.; and (iv) characterization of unimolecular reaction mechanisms by time-of-flight (TOF) measurements and velocity map imaging of photofragments and photoelectrons. A good introduction to the use of these techniques in our group can be found in our published work on diazomethane and diazirine.
The role of hydroxyalkyl radical intermediates in combustion reactions has recently been highlighted by the discovery that one of their dissociation products, the enol tautomer of aldehyde, is a significant combustion intermediate. For example, vinyl alcohol (ethenol), the less stable tautomer of acetaldehyde, is formed in flames of ethanol, olefins, and commercial fuels. Hydroxyalkyl radicals are intermediates in the reactions that give rise to enols. Diradicals are important in synthetic chemistry and photochemistry, and play a role in atmospheric, combustion and interstellar chemistry. Although carbene diradicals have been implicated as intermediates in combustion reactions, little is known about the photochemistry of the triplet ground state of the methylene diradical.
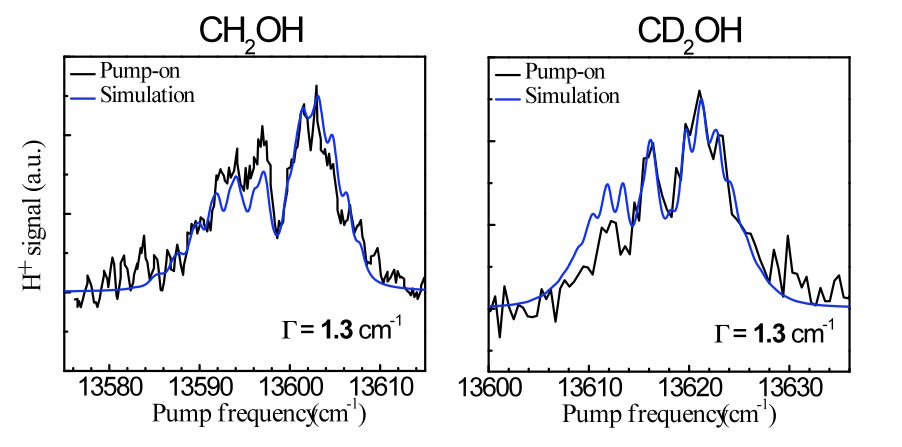
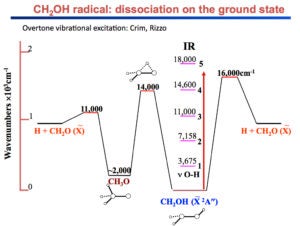
Current projects include: (i) The unimolecular reaction of CH2OH on the ground state initiated by overtone excitation to above the barriers to dissociation to CH2O + H and CH2OH → CH3O isomerization. The unique aspect of this study is that we excite directly the predissociation coordinate – the OH stretch. (ii) The photochemistry of the 2-hydroxyethyl radical, CH2CH2OH, a primary intermediate in the OH + C2H4 reaction. As this radical is not well characterized, we are carrying out a comprehensive experimental and theoretical study of its spectroscopy, excited electronic states, and decomposition and photoionization pathways. Special emphasis is placed on the role of isomerization channels. (iii) The photodissociation of triplet methylene with particular emphasis on conical intersections among Rydberg and valence states and comparisons with available high-level electronic structure calculations.
A cornerstone of our program is the synergistic confluence of theory and experiment. The iOpenShell Center directed by Professor Anna Krylov has facilitated this collaboration and students and postdocs who are interested in both theory and experiment benefit greatly from this collaboration. Theory helps interpret our experimental results and also guides the design of new experiments.
A nice example is our recent discovery of the first “roaming” mechanism in free radical dissociation. In the course of identifying products in the predissociation of CH2CH2OH and calculating branching ratios we discovered that, surprisingly, one of the product channels is H2O + C2H3. The theoretical calculations identified an unusual mechanism for this channel. Sometimes the dissociation to OH + C2H4 is frustrated and the OH radical “roams” around the ethylene fragment and abstracts a hydrogen to form water and vinyl. This is the lowest energy route to these products. We are now planning experiments to investigate this intriguing reaction. An in-depth summary of our collaborative work with the Krylov group can be found in a recent review.
3. Amorphous solid water (ASW): Volcano eruptions, phase explosions, nanobubbles
Amorphous ice is the most abundant form of water in the universe. It is important in atmospheric chemistry, e.g. in Arctic and Antarctic polar ices, clathrate hydrates, stratospheric clouds, and the Antarctic ozone hole. It is just as important in astrochemistry, where it is found in planetary rings (e.g. Saturn), comet tails, and interstellar clouds.
An ice volcano was observed, for example, on Triton (Neptune’s satellite). The eruption was observed to shoot a towering jet of material to a height of five miles, while the nitrogen atmosphere carried the smoky plume over 80 miles downwind. The ejected plume is believed to be a combination of liquid nitrogen, dust, and methane compounds driven by seasonal heating from the sun.
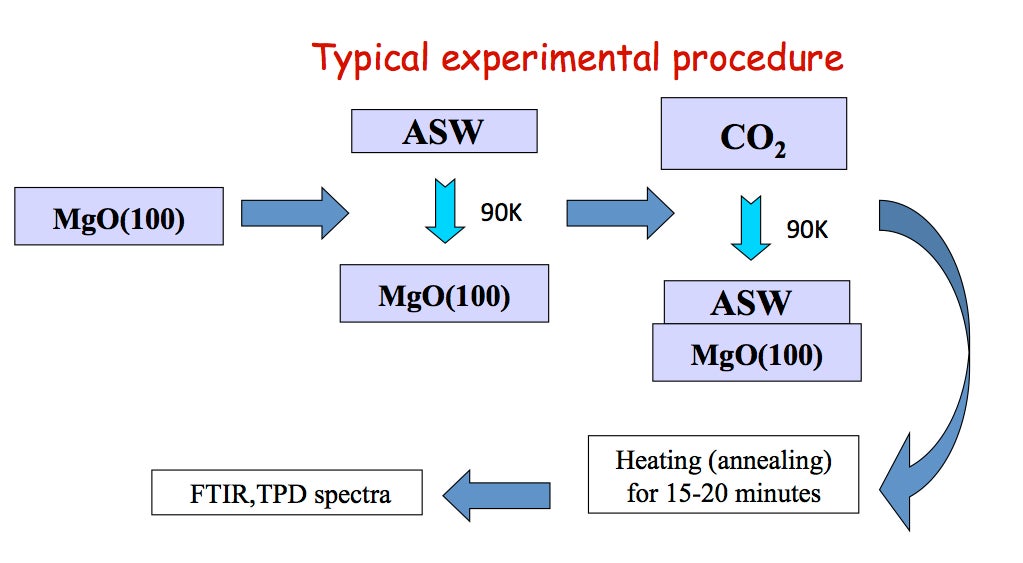
Our UHV apparatus features transmission FTIR, TPD (temperature programmed desorption), and TOF (time of flight) spectroscopy. The ice sample is deposited on MgO(100) and the temperature can be varied from ~90 to 500 K. FTIR scans of the entire mid IR region are obtained in minutes with high sensitivity (10-4-10-5 absorbance). TOF traces are taken at 200 Hz repetition rate.
In our first experiments CO2 was deposited onto ASW at 90 K. Upon annealing, some of the CO2 is trapped in cavities in the ice but the amount of trapped CO2 does not depend on CO2 dosage. The trapped CO2 is released abruptly during the phase transition from amorphous to cubic ice (~ 160 K), in what is known as the “volcano peak”. There are some CO2 molecules that remain in the ice film even after the phase transition. These molecules desorb together with H2O but their structure within the ice is unknown (Grain boundaries? Clathrates?)
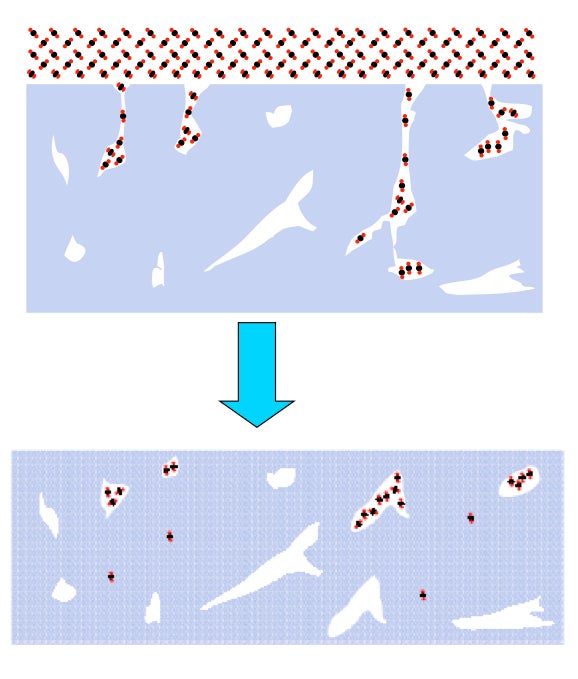
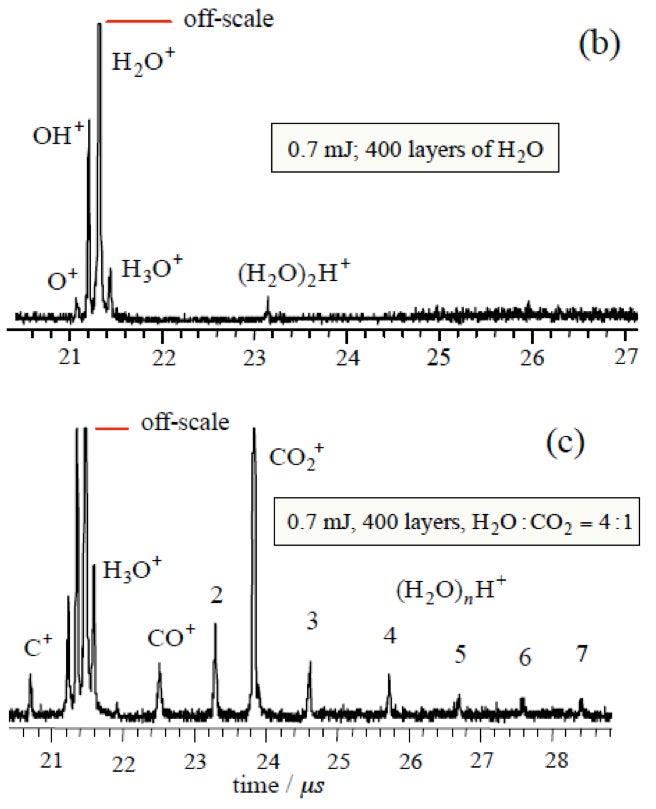
Current experiments involve TOF spectroscopic studies of laser-induced gas release from ASW. OH-stretch vibration in ASW is excited resonantly by 3425 cm-1 laser radiation. Irradiation reaches the substrate but a temperature gradient exists in the 150-500 monolayer ASW-CO2 (3:1) films. Molecules move through the detection region on time scales ~ 200 μs with lighter species passing through the detection region faster than slower ones. TOF mass spectra of ejected material are recorded at 5 μs intervals. The diameter of the IR beam at the surface exceeds greatly the film thickness, so heat transfer takes place into the substrate rather than laterally. The short time scale creates a non-equilibrium situation with consequent phase explosion and ejection of clusters from topmost layers.
When ASW is irradiated at low energies (< 0.7 mJ), only monomer water is ejected and there is no signal from protonated water clusters. However, when CO2 is added in co-deposition to ASW, with all other conditions kept the same, protonated water clusters are released in addition to monomer CO2. The CO2 has promoted a phase explosion! Our current hypothesis is that CO2 and H2O segregate and CO2 nanobubbles force material abruptly into the gas phase. These experiments are continuing and expanding.