Of Plants, Flies and Men
A torrent of fluorescent light rushes out onto the cement floor as Marie-Stanislas Remigereau opens the door to the growth chamber. Inside are hundreds of small plants precisely arranged in black trays that flank both walls up to the ceiling. Some are sprouts that have just pierced through the soil’s surface, while others have formed rosettes of green or purplish leaves. A number of plants have long thin stems with tiny white flowers and are enclosed in tall plastic cylinders.
“Look. This one reminds me of a Christmas tree,” she says over the incessant drone of the chamber’s cooling system.
Remigereau, a postdoctoral research associate in USC College, notes the variety of appearances even among plants at similar stages of growth. Rosette size, flowering time, branch number and bud size — these are physical manifestations or phenotypes of the plant’s genetic blueprint or genotype. While each plant’s genes provide the potential for the development of these physical characteristics, this is often affected by interactions with other genes and with the environment.
In other words, genes, which control the plant’s hereditary information, are a starting point for determining its structure and function. However, the route to the expression of those observable traits is highly complex, involving many interacting biochemical pathways that are yet to be fully explained.
Enter a team of scientists led by USC College’s Simon Tavaré.

In addition to serving as the principal investigator of the Center of Excellence in Genomic Science at USC, statistician-biologist Simon Tavaré and his group are also developing computational “tracking” methods to automate the observation of Drosophila melanogaster (fruit fly) behavior. Photo credit Phil Channing.
With the support of a $12.1 million, five-year grant renewal from the National Human Genome Research Institute (NHGRI), Tavaré and his colleagues are working to develop an intellectual framework, together with computational and statistical analysis tools, for illuminating the route from genotype to phenotype. Along with Arabidopsis thaliana, the small flowering plants Remigereau introduced, the other model organism that anchors their investigation is Drosophila melanogaster, a fruit fly.
“DNA is common to all organisms,” said Tavaré, holder of the George and Louise Kawamoto Chair in Biological Sciences, and research professor of biological sciences and mathematics. “So what you learn about its action in one organism in principle can be applied to others and that’s the aim here. We’re trying to understand methods with which you might tease apart how you get from genotype to phenotype in Arabidopsis and Drosophila with the expectation that those same methods should work to figure out phenotypes such as disease states in humans.”
From molecular biologists to computer scientists and genetic epidemiologists to mathematicians, a wide range of researchers from the College and the Keck School of Medicine of USC have joined Tavaré to draw a coherent, unified picture of how different genetic variants fit together and ultimately reveal the origins of human disease.
The Center of Excellence in Genomic Science (CEGS) at USC was established in 2003 with an $18.7 million grant from the NHGRI, the arm of the National Institutes of Health dedicated to advancing human health through genetic research. Since 2001, the CEGS program has funded 10 centers nationally, including those at Harvard University, Johns Hopkins University, Stanford University, Yale University and the California Institute of Technology. The goal of each center is to assemble interdisciplinary teams dedicated to making critical advances in genomic research.
From 2003 to 2008, under the direction of University Professor Michael Waterman, the CEGS at USC focused on the human genome and understanding the structure of its haplotypes, the common strings of DNA that are passed through generations.
The typical human genome is composed of two sets of threadlike DNA-containing structures called chromosomes, one set inherited from the mother and the other from the father. With the completion of the Human Genome Project in 2003 and the ability to identify mutations in DNA, researchers now have a set of tools that make it possible to find the genetic contributions to common diseases. One approach to identifying genes involved in human disease is genome-wide association studies.
This relatively new statistical method seeks to correlate the occurrence of variations at the DNA level (mutations) with differences in a phenotypic trait (a diseased state in humans, or the general appearance of each fly or plant). Association methods allow researchers rapidly to scan the genome for small variations, called single nucleotide polymorphisms or SNPs (pronounced “snips”), that occur more frequently in people with a particular disease than in people without the disease. Scientists then use data gathered through association studies, which can examine hundreds of thousands of SNPs at the same time, to pinpoint genes that may contribute to a person’s risk of developing a certain disease.
During the first five years of the CEGS grant, Waterman, who is also USC Associates Chair in Natural Sciences and professor of biological sciences, computer science and mathematics in the College, and his collaborators helped pioneer improved techniques for conducting association studies that are now commonly used by the biological community.
With the CEGS grant renewal, Tavaré and his colleagues are building upon these advances with some novel experimental approaches. Rather than focusing on single SNPs, recently developed technologies will now enable them to sequence whole genomes and find all the positions at which any two individuals differ in their DNA sequences. The group is also measuring intermediate phenotypes, such as the expression of each individual gene in specific tissues, to refine association tests and scale them up to the whole genome level.

Postdoctoral researcher Marie-Stanislas Remigereau and third-year molecular biology doctoral student Pei Zhang discuss Arabidopsis thaliana that are cultivated in two growth chambers in the basement of Ray R. Irani Hall. Photo credit Max S. Gerber.
In Drosophila, they are using previously identified DNA sequences to create genetically identical model organisms that allow them to more directly observe what happens to phenotypes under differing conditions. Since Arabidopsis is a naturally inbred species with each plant producing an offspring with an identical genetic make-up, researchers are able to track exactly which changes in the plant’s physical appearance are responses to different climates.
“Understanding how you get from genotype to phenotype is arguably one of the biggest problems in biology at the moment and will probably remain so for a long time,” Tavaré said. “Because an enormous amount is known about the genetics of these two model organisms, I think we have an advantage over trying to do this directly in humans.”
Arabidopsis and Drosophila are an ideal fit for these studies primarily due to the size of their genomes and because they share a number of genes in common with humans that are known to be linked to disease.
In 2000, Arabidopsis was the first flowering plant genome to be sequenced and it contains about 25,500 genes, which is close to the lower estimates for the number of genes in the human genome. Approximately 100 Arabidopsis genes are similar to disease-causing genes in humans, including the genes for breast cancer and cystic fibrosis.
Associate Professor of Biological Sciences Magnus Nordborg and his team including Remigereau and Richard Clark, assistant professor of biology at the University of Utah, are looking at roughly 150 different phenotypes in Arabidopsis. Among them is flowering time, which is most indicative of the plant’s overall adaptation to climate and environmental conditions in general.
“Arabidopsis is arguably the best model organism for dissecting the genotype-phenotype map,” Nordborg said. “It naturally exists as inbred lines that are genetically adapted to a wide variety of environments, and can readily be grown in large numbers under controlled conditions.
“By 2014, we hope to have identified the major determinants of flowering time variation in Arabidopsis as well as have a good overall picture of how these variants, together with the environment, lead to variation at the phenotypic level.”
Drosophila has been used as a model organism in genetics for more than 100 years and its genome sequence was first reported in 2000. With 13,700 genes, Drosophila has homologues for many genes known to be involved with human disease, including cancer.
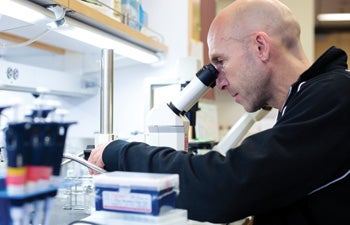
Professor of Biological Sciences John Tower uses a stereo microscope to score genetic markers in Drosophila melanogaster (fruit fly). Photo credit Max S. Gerber.
Professor of Biological Sciences Sergey Nuzhdin, Gabilan Assistant Professor of Biological Sciences Michelle Arbeitman and Professor of Biological Sciences John Tower have joined forces to probe such phenotypes as aging and courtship behavior in Drosophila and discover how each presents under different conditions. Tavaré’s lab is also developing computational “tracking” methods to automate the observation process.
“We are starting to understand which regulatory polymorphisms contribute to the differences in transcription among individuals,” Nuzhdin said of the group’s progress thus far. “Flies are an important model organism to look for insights as their regulatory polymorphisms segregate independently of one another in natural fly populations, while in humans these polymorphisms are organized into linkage-disequilibrium blocks and causal polymorphisms are nearly impossible to identify.
“Together John and Michelle’s labs along with mine, in close collaboration with our colleagues in computational biology and at the Keck School, are now beginning to apply our approaches to the whole genome at the same time.”
Experimental results from each group will be combined with sequence data generated by the USC Epigenome Center located in the USC Norris Comprehensive Cancer Center. The nation’s first such center, established in 2007 and directed by Professor Peter Laird, is a state-of-the-art genomics laboratory with expansive, dedicated bioinformatics facilities.
Next, quantitative analyses of the resulting data will be performed by Associate Professor of Biological Sciences and Computer Science Ting Chen, Assistant Professor of Biological Sciences Liang Chen, Assistant Professor of Biological Sciences Andrew Smith, Professor of Biological Sciences Fengzhu Sun, and Associate Professor of Biological Sciences Xianghong Jasmine Zhou.

Professor of Biological Sciences Sergey Nuzhdin (center) and members of his lab — Bradley Main, Margaret Sherriffs, Yu Huang (of Associate Professor of Biological Sciences Magnus Nordborg’s lab), Julia Saltz, Courtney Fjeldsted, and Maren Friesen — investigate genetic variation. Photo credit Max S. Gerber.
Together this team of computational biologists will develop novel, efficient algorithms that will allow them to scan enormous amounts of sequencing data generated through the Epigenome Center and better identify the possible SNPs or genetic variant sets that are responsible for the phenotypic expressions observed in Arabidopsis and Drosophila.
“One of the biggest challenges we face is improving and enforcing statistical methods of quality control such as accounting for false positives,” Liang Chen said. “In an effort to best enforce quality control, we consider intermediate phenotypes, which although not directly related provide extra information about the path from genotype to phenotype.”
Once these new methods for understanding the genotype-to-phenotype map have been developed in the first three years of the grant, Tavaré and his USC College colleagues will partner with preventive medicine faculty members at the Keck School to investigate how these techniques might be applied to existing human cancer studies.
Keck School’s Paul Marjoram, Kimberly Siegmund and David Conti point out that while phenotypes might be different across organisms, they are often measured on the same scale, so techniques developed for one organism’s phenotype can be applied to the analysis of another.
“There is the real prospect that using the model systems in CEGS will help us refine and verify methods we develop for analyzing human data,” Marjoram said. “This gives us the experience we need to more successfully apply them to human data and more accurately interpret these new results when looking at colon cancer data, for example.”
Marjoram and his colleagues plan to begin by applying their findings to the Colon Cancer Family Registry, which is supported by National Cancer Institute and to date includes data and biospecimens for more than 30,000 subjects from multiple institutes.
“The answers to many questions are written in our DNA,” he said. “Our hope is to develop methods that allow us to uncover as many of these answers as we can, as quickly as we can.”

Steven Finkel, deputy director of the Center for Excellence in Genomic Science at USC and director of its Minority Action Plan, with molecular and computational biology doctoral students Lorraine Provencio and Eduardo Ayala. Photo credit Laurie Moore.
The Center of Excellence in Genomic Science Minority Action Plan at USC
Not only does the Center of Excellence in Genomic Science (CEGS) at USC carry out its research mission, it also administers a National Institutes of Health-funded Minority Action Plan (MAP), which provides education and training about basic research in the life sciences to under-represented minorities.
“Many students don’t know what it’s like to be a scientist,” said Steven Finkel, associate professor of biological sciences as well as deputy director of the CEGS at USC and director of its MAP program. “So the goal of our program is to give, particularly undergraduates, the opportunity to conduct high-quality research under the mentorship of a faculty member as well as guide students through their educational and career transitions.”
The CEGS at USC, which initially began in 2003, runs three programs: an academic-year research program for undergrads called the Genomics Research Experience for Undergraduates (GREU); a summer version of GREU, which includes a journal club and seminar series; and the Genomics Graduate Scholars (GGS) program, which provides stipends for minority graduate students working in laboratories in the biological sciences.
Approximately $1.5 million of the CEGS grant renewal is directed to the MAP program at USC. This generous funding currently supports the highest number of participants yet: 19 undergrads and seven doctoral students.
Read more articles from USC College Magazine’s Spring/Summer 2010 issue.